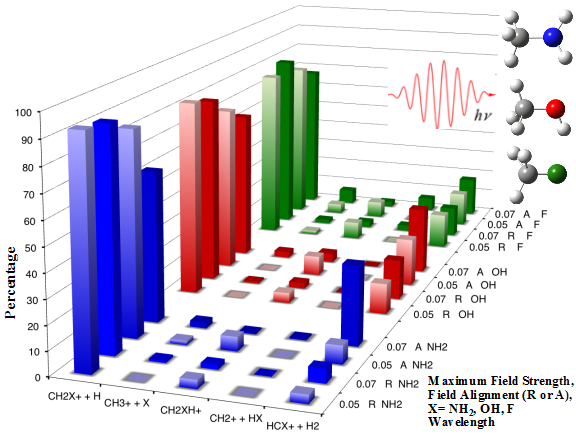
Molecular dynamics can provide a detailed description of the behavior of molecules during reactions. In ab initio molecular dynamics (AIMD), whenever information about the potential energy surface is needed for integrating the equations of motion, it is computed “on the fly” using electronic structure calculations. In Born-Oppenheimer molecular dynamics (BOMD), the electronic structure calculations are converged, whereas in the extended Lagrangian approach the electronic structure is propagated along with the nuclei. We have developed a Hessian-based predictor-corrector algorithm for the efficient computation of Born-Oppenheimer ab initio molecular dynamics, and the atom centered density matrix (ADMP) approach for extended Lagrangian classical trajectory calculations of molecular systems. We have applied these methods to study a wide range of reactions.
a. Bakken, V.; Millam, J. M.; Schlegel, H. B.; Ab Initio Classical Trajectories on the Born-Oppenheimer Surface: Updating Methods for Hessian-Based Integrators. J. Chem. Phys. 1999, 111, 8773-77 (10.1063/1.480224).
b. Schlegel, H. B.; Millam, J. M.; Iyengar, S. S.; Voth, G. A.; Daniels, A. D.; Scuseria, G. E.; Frisch, M. J.; Ab Initio Molecular Dynamics: Propagating the Density Matrix with Gaussian Orbitals. J. Chem. Phys. 2001, 114, 9758-63 (10.1063/1.1372182).
c. Rega, N.; Iyengar, S. S.; Voth, G. A.; Schlegel, H. B.; Vreven, T.; Frisch, M. J.; Hybrid Ab Initio / Empirical Molecular Dynamics: Combining the ONIOM Scheme with the Atom-Centered Density Matrix Propagation (ADMP) Approach. J. Phys. Chem. B 2004, 108, 4210-4220 (10.1021/jp0370829).
d. Schlegel, H. B.; Molecular dynamics in strong laser fields: a new algorithm for ab initio classical trajectories J. Chem. Theory Comput. 2013, 9, 3293–98 (10.1021/ct400388j).